
Background: how we got here
CROs emerged when sponsors struggled to keep all trial operations in-house while regulations, documentation, and global site footprints exploded. The outsourcing model promised specialist expertise, flexible staffing, and scalability without permanent headcount expansion.
Regulatory requirements continuously expanded — with additional safety reporting and monitoring expectations — driving increased CRO adoption across oncology and complex therapeutic areas, encompassing feasibility through pharmacovigilance. Consequently, trials are often designed around what a large CRO can operationalise, rather than what is simplest and most informative for patients.
The current problem: bloated costs and bureaucracy
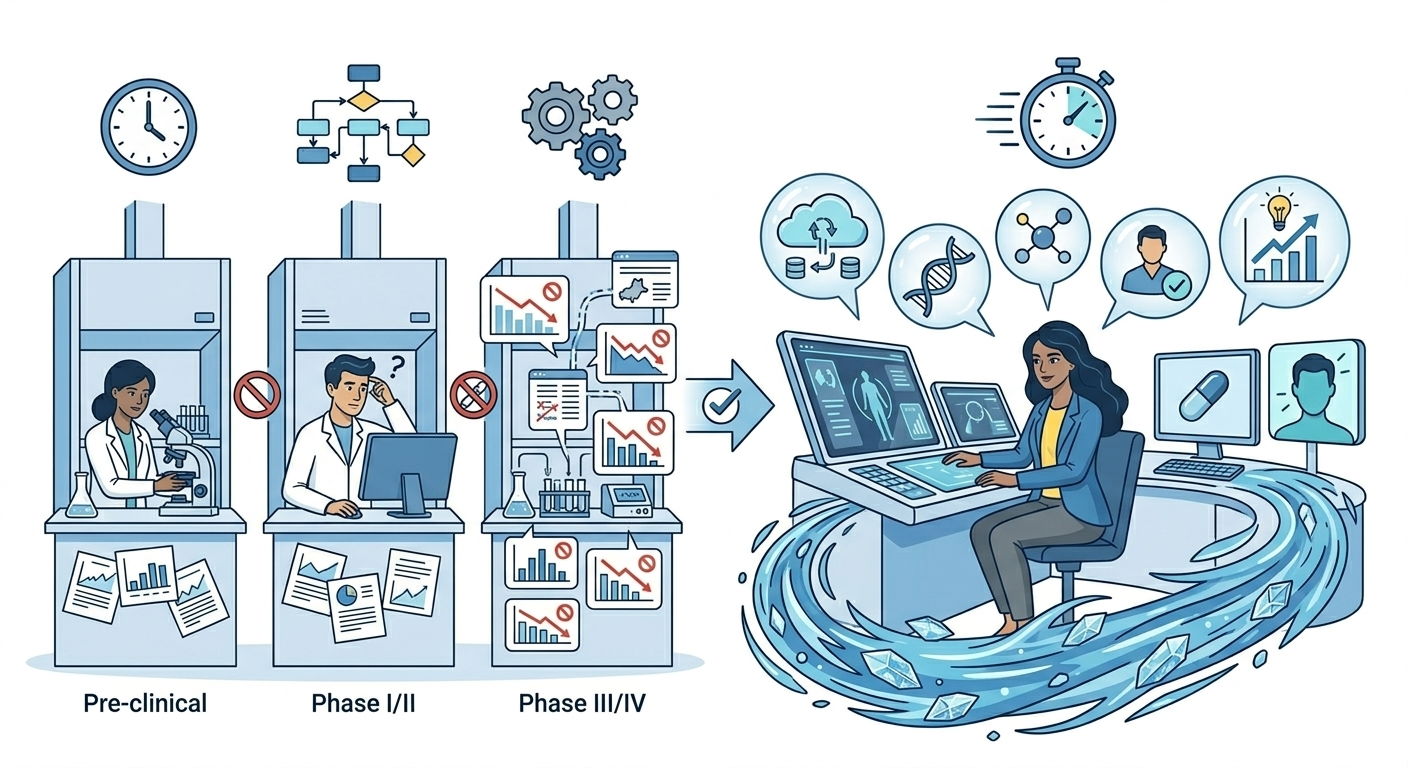
Many stakeholders view CROs less as partners and more as large machines optimised for selling hours, not for eliminating waste. Three recurring issues emerge:
- Layers of bureaucracy: Each outsourced function introduces separate templates, trackers, and reviews. Investigators receive excessive low-value queries and redundant forms driven by process requirements rather than scientific necessity. Administrative overhead becomes disproportionate for low-risk pragmatic trials.
- Bloated costs and opaque mark-ups: Multiple organisational layers generate overhead and margins. Change orders, complex feasibility work, and rework consume budgets without improving data quality. Sponsors subsidise expensive infrastructure for relatively simple study designs.
- Misaligned incentives: Revenue tied to staffing creates minimal motivation to streamline operations. Effort concentrates on easily quantifiable metrics rather than meaningful outcomes like answer speed or patient experience. Data-driven approaches remain theoretical rather than operationalised.
The result: slower study start-up, overburdened investigators, and rising trial costs without proportional gains in scientific value.
The solution: lean, data-first alternatives
Rather than organising around logistics and headcount, a healthier model is built around questions and data. The right question to ask is: what is the minimum system we need to generate robust, decision-grade evidence?
Key principles include:
- Start from the decision, not the org chart: Define the regulatory, payer, or clinical decisions required, then identify the minimal endpoints and data sources necessary. Incorporate real-world data, ePROs, and digital measures to reduce site burden.
- Strip away unnecessary bureaucracy: Apply risk-proportionate processes — preventing low-risk studies from receiving high-risk trial treatment. Standardise essential elements while eliminating duplicate forms and approvals.
- Use small, expert teams instead of layers: Replace multiple management tiers with compact interdisciplinary teams combining scientific, operational, and data expertise — minimising miscommunication through proximity.
- Let technology remove work, not add it: Deploy eConsent, ePRO, wearables, and remote monitoring to reduce visits and manual transcription. Use real-time dashboards for targeted rather than blanket monitoring.
- Transparent, value-based pricing: Align budgets to outcomes achieved rather than FTE capacity. Clearly separate pass-through costs from mark-ups.
What this means in practice
For sponsors, the benefits are tangible: faster set-up, clearer lines of communication, and more budget going into science and data rather than overhead.
As regulatory frameworks mature, the industry is moving away from logistics-heavy models toward lean ecosystems where expert teams, smart technology, and proportionate governance deliver the evidence that actually changes practice.
This is the model Comet Clinical was built on. We believe great clinical research should be defined by the quality of the evidence it generates — not the complexity of the machinery required to produce it.